Primary ciliary dyskinesia (PCD) is a rare, genetic disorder of the motile cilia characterised by chronic lung disease, nasal and sinus disease, chronic ear infections with glue ear leading to possible hearing impairment, and subfertility. Half of patients have organ laterality defects. Despite symptoms being present from birth, PCD remains underdiagnosed. Early diagnosis and symptom management may reduce morbidity, improve quality of life and ensure children are adequately supported to meet their full potential.
This article seeks to raise awareness of PCD in children's nurses who are well placed to identify children for referral to the national PCD diagnostic service. It examines the clinical characteristics of the disease along with the complexities and limitations of diagnostic testing. In addition, it explores the management of children with PCD within the national PCD management service for children.
Nursing Children and Young People. doi: 10.7748/ncyp.2017.e936
Correspondence Peer reviewThis article has been subject to open peer review and has been checked for plagiarism using automated software
Received: 05 April 2017
Accepted: 15 June 2017
Published online: 21 August 2017
This article aims to raise awareness of primary ciliary dyskinesia (PCD), a rare inherited condition caused by abnormal cilia, which is often underdiagnosed due to lack of awareness among healthcare professionals (Behan et al 2016a). It outlines clinical symptoms, diagnostic methods and management, and highlights which patients warrant investigations by the national PCD diagnostic service.
After reading this article and completing the time out activities, you should be able to:
• Describe the main symptoms and presentation of PCD and how screening may identify children to be referred for diagnostic testing.
• Consider the effect of PCD on children and their families as a genetic, lifelong disease.
• Outline the importance of adequate transition of young people with PCD, especially given the lack of a commissioned adult service.
• Identify children for further investigation.
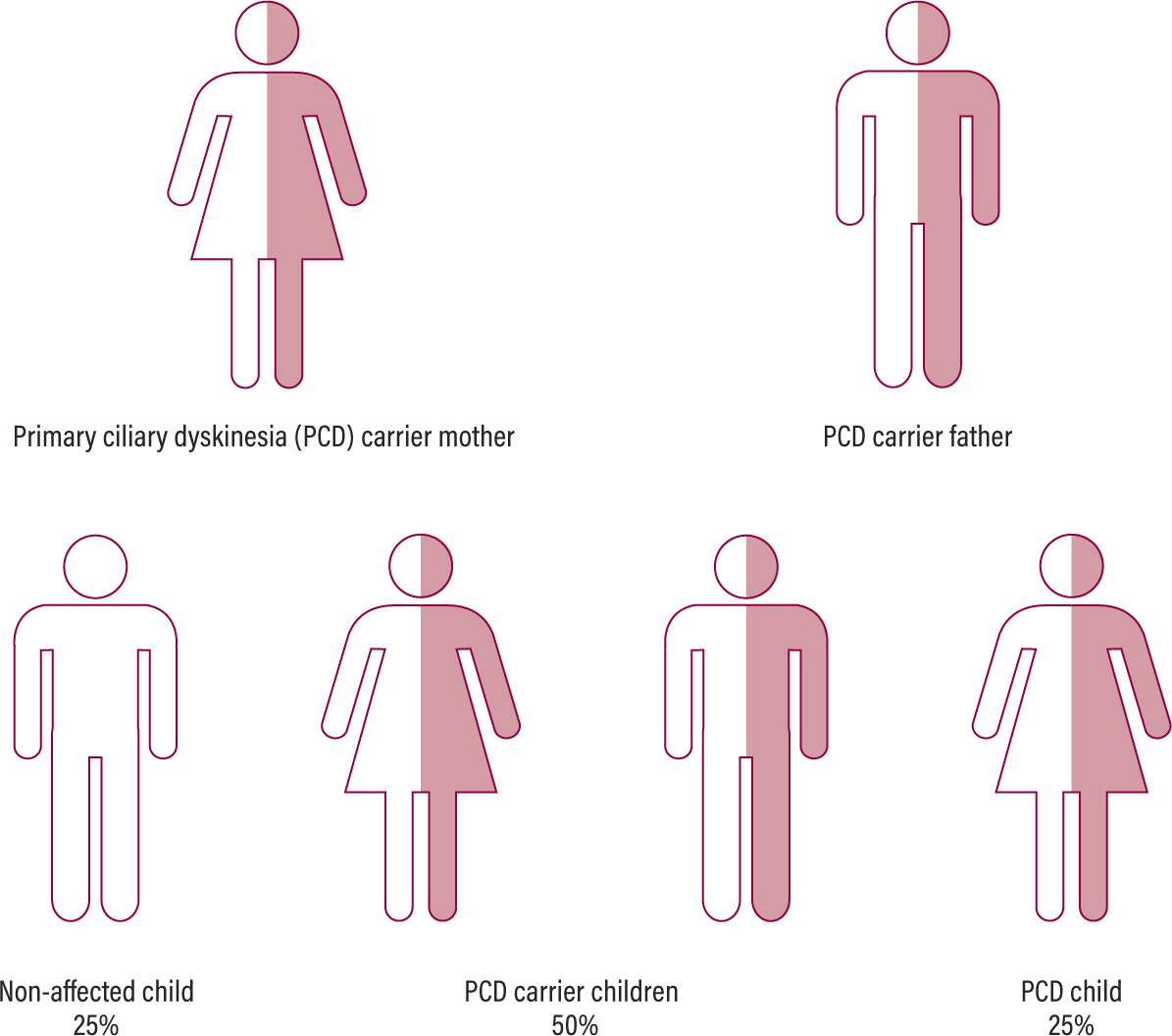
PCD is a rare, autosomal recessive disorder, meaning that an affected gene must be inherited from each parent (Figure 1). In patients with PCD the structure and function of motile cilia (microscopic, hair-like structures) throughout the body are affected. Ciliated epithelium lines the lungs, nasal and sinus cavities and Eustachian tubes in the ear, as well as the fallopian tubes (Figure 2). The tail of a sperm shares a similar ultrastructure to cilia (Barbato et al 2009).
Cilia form the basis of the mucociliary escalator clearing the airways of mucus and debris as part of the body's defence system. Mucus is secreted by goblet cells in the airways to trap bacteria and debris. Cilia beat in a coordinated manner to move the mucus to the back of the throat where it is either swallowed or expectorated. In PCD, cilia are either static, ineffective or absent, leading to a build-up of mucus in the chest, ears, nose and sinuses. Symptoms occur from the neonatal period onwards and include (Goutaki et al 2016):
• Daily chronic wet cough.
• Recurrent chest infections leading to bronchiectasis (irreversible lung damage).
• Chronic rhinitis and rhinorrhoea.
• Chronic and recurrent sinusitis.
• Recurrent otitis media.
• Glue ear leading to hearing loss.
• Subfertility.
• Around 50% of patients have dextrocardia (heart on the right) and/or situs inversus totalis (mirror image arrangement of organs) (Figure 3).
• Around 5% of patients have other laterality defects or heterotaxy (unusual arrangement of internal organs), which can cause complex congenital cardiac disease, gut malrotation, asplenia or polysplenia.
The prevalence of PCD is unknown due to the complexity of diagnosis and lack of awareness among healthcare professionals. The literature shows a wide range of estimates varying from 1:2,200 in consanguineous populations (where interfamily marriage is common) to 1:40,000. Expert consensus estimates the likely prevalence is 1:10,000 live births. For comparison, cystic fibrosis has an incidence of 1:2,500, approximately four times more common (Lucas et al 2014, Goutaki et al 2016).
Go to the University Hospital Southampton NHS Foundation Trust What is PCD? website and watch the videos of motile and immotile cilia: uhs.nhs.uk/OurServices/Childhealth/Primary-ciliary-dyskinesia/what-is-pcd.aspx
Since 2006 NHS England has commissioned a national diagnostic service for PCD. Adults and children can be referred to one of three centres in Leicester, London or Southampton. PCD is complex to diagnose; it involves several expensive tests each requiring expertise to conduct and interpret the results (Lucas et al 2017).
There is no gold standard test to diagnose PCD and each investigation has limitations. Diagnosis involves:
• Detailed clinical history.
• Nasal nitric oxide (nNO) measurement.
• Nasal brushing to harvest ciliated epithelium for:
PCD symptoms are non-specific and many ‘healthy’ children have frequent respiratory infections, rhinitis, ear infections or glue ear (Goutaki et al 2016). This makes it difficult to identify who needs referral for diagnostic testing. An international patient and parent survey undertaken by Behan et al (2016b) highlighted that, before diagnosis, parental concerns were not taken seriously by medical professionals. By the time a child was diagnosed they had undergone unnecessary ear surgery, had multiple courses of intravenous antibiotics and, in some cases, already had bronchiectasis. Early diagnosis and management of the condition may improve patient outcomes and reduce morbidity (Noone et al 2004).
Behan et al (2016a) reviewed 641 consecutive patients referred for PCD diagnostics at two UK centres. They looked at the range of symptoms experienced by patients in PCD-positive and PCD-negative groups and devised a simple predictive tool, PICADAR, to help clinicians identify which patients warrant further testing in specialist centres. A summary of symptoms is shown in Table 1.
| History and examination | % |
|---|---|
| Persistent wet cough | 97 |
| Rhinitis | 81 |
| Neonatal respiratory distress | 74 |
| Ear symptoms | 61 |
| Hearing loss | 48 |
| Dextrocardia | 46 |
(Adapted from Behan et al 2016a)
Go to erj.ersjournals.com/content/early/2016/02/25/13993003.01551-2015.figures-only and look at the PICADAR tool (Figure 2). The tool should only be used in patients with a daily wet cough. Think of patients you have seen in your practice and apply the tool to them. What do you think are the merits of using such a tool? Are there any limitations of using such a scoring system?
Respiratory distress affects up to 87% of neonates with PCD (Goutaki et al 2016). Mullowney et al (2014) conducted a study to identify characteristics that differentiate infantile respiratory distress in those with and without PCD. They concluded that PCD should be considered in infants with unexplained respiratory distress if situs inversus, lobar collapse or prolonged oxygen therapy is also seen. They reported a later onset of respiratory distress in the PCD group, a median of 12 hours compared with one hour in non-PCD infants.
In PCD there is no effective mucociliary clearance. Mucus is cleared only with physiotherapy or cough. In cystic fibrosis, a wet cough indicates infection, but children with PCD always have a wet cough even when they are well, making infections difficult to diagnose (Strippoli et al 2012).
PCD patients often have chronic nasal congestion and rhinitis that does not clear with antihistamines or other treatments. Many children experience recurrent sinusitis.
In the ears, mucociliary clearance in the eustachian tubes is responsible for clearing the middle ear. Children experience recurrent otitis media, glue ear and hearing loss. Unlike unaffected children with glue ear, grommet insertion is often unsuccessful and can result in offensive discharge and no improvement in hearing.
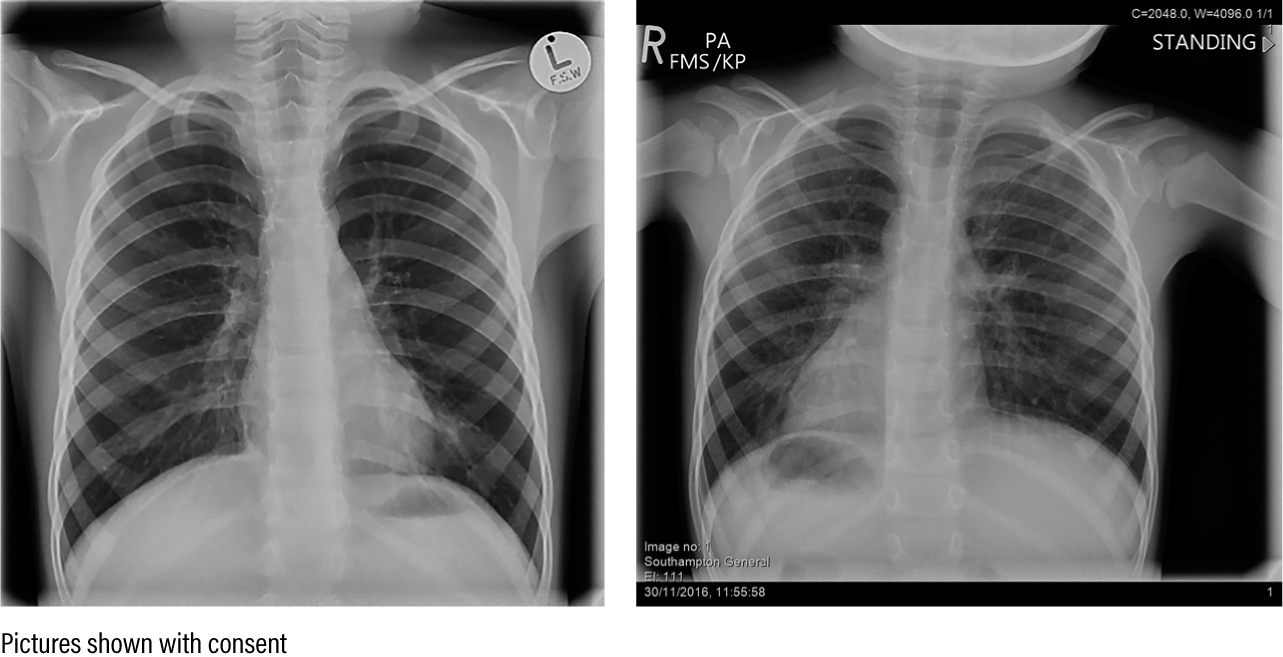
Embryonic nodal cilia are involved in the lateral formation of the organs. In PCD there is a 50:50 chance of situs abnormalities. Situs inversus does not cause problems; Figure 3 shows chest X-rays of two PCD patients, one with normal situs and the other with situs inversus. However, approximately 5% of children with PCD have other organ arrangements (situs ambiguous or heterotaxy), which can result in congenital heart defects, asplenia, polysplenia and malrotation of the gut (Shapiro et al 2016).
For reasons that are not understood, most patients with PCD have low nNO. Levels can be measured using a variety of methods, for example, exhale against resistance and tidal breathing, and instruments, such as portable, hand-held electrochemical devices (Figure 4) and ‘gold standard’ chemiluminescence analysers (Figure 5). International guidelines recommend using a chemiluminescence device with a velum closure manoeuvre (American Thoracic Society and European Respiratory Society (ERS) 2005). Young children are often unable to achieve this, in which case tidal breathing sampling suffices, but results must be interpreted with caution (Lucas et al 2017).
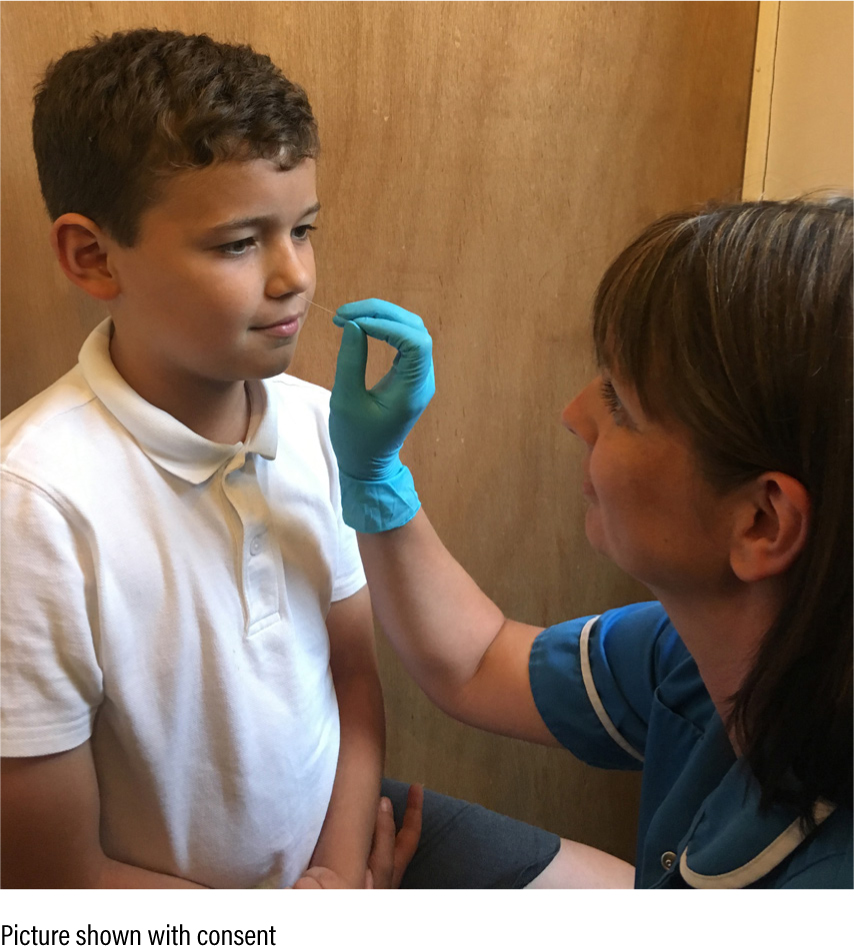
Ciliated cells are most easily sampled from the nose. A cytology brush is inserted into the nostril and agitated along the turbinate (Figure 6). Sampling is uncomfortable but not painful, and takes only a few seconds. Viral or bacterial infections can cause abnormal ciliary function that is difficult to differentiate from PCD, and patients should therefore be well for four to six weeks before sampling (Lucas et al 2017).
High-speed video microscopy is used to assess ciliary beat frequency and pattern. The live sample is observed and recorded. Images are played back at a slow rate for analysis. If the cilia have a normal beat pattern, and the nNO was within normal limits, the diagnosis of PCD is extremely unlikely and further testing is rarely warranted.
Abnormal high-speed video microscopy alone is not enough to confirm PCD. Patients often have secondary ciliary defects that can be difficult to differentiate from PCD. Therefore, if abnormalities are seen further laboratory assessments are needed (Lucas et al 2017).
Nasal brushing cells are cultured in an incubator with the cells fed nutrients and antibiotics from below. New cells develop cilia on the surface exposed to air. These cilia are free from secondary defects, so if abnormalities persist when reanalysed by high-speed video microscopy the defect is primary (genetic), that is, PCD. The culturing process is technically difficult and is only successful in 50-60% of cases (Hirst et al 2014).
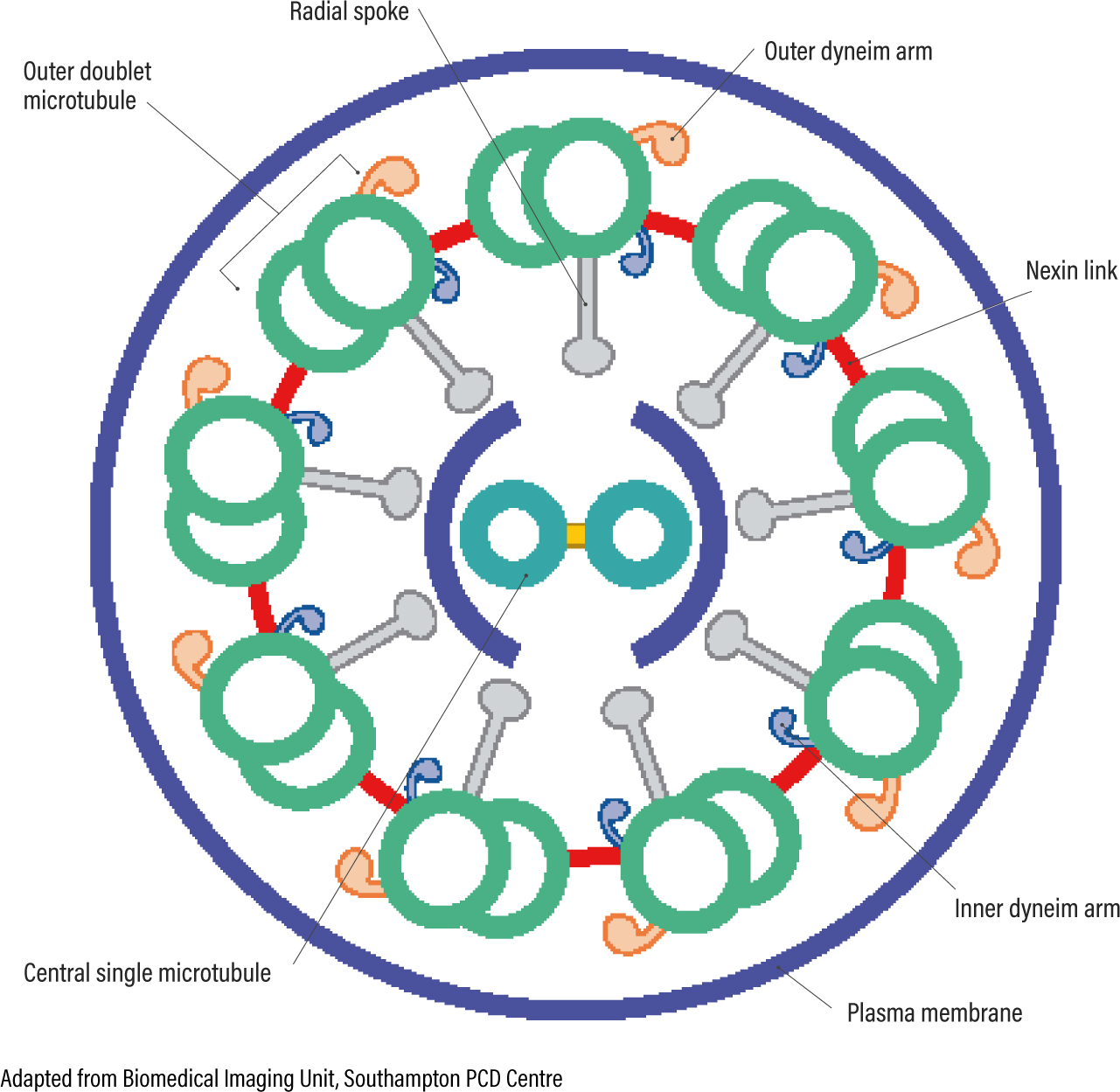
Using transmission electron microscopy, the axonemal ultrastructure (internal cross section) of the cilia is examined. Preparation and analysis of the sample is complicated, time consuming and highly specialised. The normal axoneme (Figure 7a, b, c) is described as a 9 + 2 arrangement with nine outer doublet microtubules surrounding two central single microtubules. Each doublet has an inner and outer dynein arm that work to power the cilia. The radial spokes and nexin links keep the structure of the axoneme and the central pair of microtubules determine the direction of travel. If any part of the structure is missing or incorrectly formed, this can affect the functionality of the cilia (Stannard et al 2010, Shoemark et al 2012). Different genes code for the different proteins that make up the components of the ultrastructural axoneme. This accounts for the heterogeneity (many disease-causing genes) of the disease (Knowles et al 2013).
Until recently, transmission electron microscopy was considered the ‘gold standard’ test for PCD. Although hallmark ultrastructural abnormalities can confirm the diagnosis, it is now known that approximately 15% of patients have normal ciliary ultrastructure, hence the need for additional tests (Lucas et al 2017).
Immunofluorescence (Figure 8) involves the staining of specific ciliary proteins with fluorescent antibodies to identify when structures in the cilia are missing. It is cheaper and quicker than transmission electron microscopy yet only identifies some defects so cannot be used as a stand-alone test (Shoemark et al 2017).
Genetic testing for PCD is expensive and complex. Unlike cystic fibrosis, which has one disease-causing gene with many possible mutations, PCD is caused by >35 genes each with many possible mutations. Known genes cause 60-75% of PCD (Davis et al 2015, Marshall et al 2015). Until more genes are discovered it is not possible to exclude the diagnosis based on genotype. However, if causative mutations are found, this can confirm the diagnosis (Lucas et al 2017). Patients with a diagnosis of PCD, who do not have a genetic cause identified, are eligible for the 100,000 Genomes Project, which aims to sequence 100,000 genomes from patients with a rare disease, plus their families and patients with cancer (Genomics England 2017).
The first evidence-based guidelines for diagnosing PCD have been published by the ERS PCD taskforce (Lucas et al 2017). The guidelines include assessment of each method contributing to the diagnostic pathway. The taskforce recommends that a definite positive diagnosis of PCD can only be made if ‘hallmark’ transmission electron microscopy defects or causative genetic mutations are identified. Since many PCD patients are likely to fall outside these parameters, the guidelines recommend that patients with a suggestive clinical history and low nNO are considered ‘highly likely’ if high-speed video microscopy is consistently abnormal or if high-speed video microscopy suggests PCD after cell culture.
Excluding PCD in patients with equivocal results is not possible. Many patients end up with an uncertain outcome, despite using state-of-the-art investigations. Genetics is a growing field and electron tomography, immunofluorescence and radioaerosol mucociliary clearance testing have the potential to improve diagnostic certainty.
Go to erj.ersjournals.com/content/early/2016/11/11/13993003.01090-2016.figures-only to look at the diagnostic pathway (Figure 4 at the link). What do you think are the implications for a child and family without a definitive diagnosis, but still dealing with the characteristics of the disease? Many families struggle to be referred in the first place given the non-specific symptoms of the disease. Once referred, they are immersed in a complicated pathway that is not understood by the wider healthcare team. Adaptations at school, antibiotics from a GP and disability benefits/free healthcare outside the UK may be much more difficult to access without a definitive diagnosis.
National diagnostic centres comply with the NHS England service specification, with each centre differing slightly in its local delivery. In Southampton, nurses and physiotherapists work across diagnostic and management services. To obtain a viable sample for analysis patients should be infection free for four to six weeks before testing. Telephone triage enables the nurse to identify patients whose appointments should be postponed, perhaps recommending antibiotic prophylaxis and/or nasal douching before clinic. Frustratingly for the patient, this can cause delays by weeks or even months, but preventing brushings which will not result in a conclusive outcome is time and cost effective for the NHS, and it prevents repeated sampling for the patient.
The nurse may be responsible for clinical history taking, measuring and interpreting nNO levels, nasal douching, nasal brushing, spirometry and collecting samples for genetics. It is vital that patients and families understand the complexities of the tests, and consequent impact on timelines before a definitive diagnosis is made. Support for families whose diagnostic journey is complex and protracted is an important part of the nursing role.
PCD centres work closely with the PCD Family Support Group. The charity is a motivated organisation comprising PCD patients and their families. It was pivotal in securing commissioning of the diagnostic and management service and remains an important partner in the national PCD service. The charity's website (pcdsupport.org.uk) contains a wealth of accurate and up-to-date information compiled by the committee in collaboration with clinicians and scientists from the service. It additionally provides telephone support and an online patient forum.
Go to the PCD Family Support Group website and watch the 20-minute video What is PCD? to consolidate the information you have read so far: pcdsupport.org.uk/what-is-pcd
Look at some of the case studies on the website to gain a better insight into how a diagnosis of a rare disease affects patients and their families.
In 2012 the national management service for PCD in children was commissioned. Alongside the three PCD diagnostic centres a fourth management centre was established in Leeds/Bradford to care for children in the north of England.
The service is commissioned to provide an equitable service for all children with PCD in England, ensuring they are managed within agreed standards of care. The aims of the service are to:
• Manage the care of children in a multidisciplinary one-stop clinic.
• Optimise lung function and reduce rate of decline.
• Manage ear, nose and throat symptoms, assess hearing and ensure aiding is used where necessary.
• Ensure an adequate transition takes place, empowering young people to manage their disease on their transfer to adult services.
Referrals to the service are made from the national diagnostic service or PCD centres in other countries. Patients' diagnostic criteria are checked and any outstanding testing requested. An echocardiogram and abdominal ultrasound are conducted to ascertain situs and associated problems.
Children are reviewed by the PCD multidisciplinary team at least annually. This includes assessment by a PCD respiratory physician, ear, nose and throat physician with an interest in PCD, PCD physiotherapist, PCD nurse and audiologist. A personalised management plan is revised in discussion with the family.
The service coordinates each child's medical care ensuring they have access to services relating to all aspects of PCD care. It provides education to healthcare professionals, patients and families and agencies involved with the child. Each centre has a telephone advice service for families and professionals.
European consensus guidelines recommend that children should be seen once every three months for respiratory assessment, for example, spirometry and sputum microbiology, with access to bronchoscopy and more comprehensive lung function testing as required (Barbato et al 2009). Chest physiotherapy twice a day is the mainstay of treatment. This can be undertaken in a variety of ways and is adapted as the child grows. All children are regularly reviewed by a physiotherapist and have access to the clinical nurse specialist for education and support.
Much of the care is managed on a case-by-case basis. Exacerbations are treated following the PCD service antibiotic protocol while those who have frequent exacerbations may be offered prophylactic antibiotics. With concerns about antimicrobial resistance, given the lack of evidence to support use, there has been a move to reduce the number of children on prophylactic antibiotics or to introduce treatment breaks in the summer months.
Respiratory infections are treated aggressively with two weeks of targeted oral antibiotics determined by microbiology results, and patients are advised to increase physiotherapy. Stubborn infections may require IV antibiotics with a small number of children requiring regular three-monthly courses. Pseudomonas aeruginosa infections are treated in line with cystic fibrosis guidelines.
Anecdotally, children benefit from the use of nebulised mucolytics to loosen secretions and aid clearance. As with all PCD care evidence remains sparse. Patients are managed on a case-by-case basis and mucolytics commenced when clear benefit is demonstrated.
Before diagnosis, many patients commence asthma treatments in a bid to control their chest symptoms, often with little or no effect (Strippoli et al 2012). Bronchodilator responsiveness (reversibility) is therefore tested and patients given a trial without asthma preventers if there is no evidence of concurrent asthma. Patients requiring inhaled corticosteroids and bronchodilators should use a spacer and have their inhaler technique checked.
A small number of children require supplemental oxygen with or without non-invasive ventilation. Occasionally, children have such extensive lung disease that they are referred for transplant assessment.
Children with PCD often have glue ear resulting in conductive hearing loss. Regular audiology is essential to assess hearing and provide hearing aids if necessary (Majithia et al 2005). Often hearing deteriorates during upper respiratory tract infections and improves in between. Good communication between schools and families is essential to ensure children reach their full potential. All children are offered a school visit to explain PCD care with special emphasis given to hearing because of its importance to education. Information for schools is available to download from the PCD Family Support Group (2017) website.
Anecdotally, most children benefit from nasal/sinus douching. This involves washing the nose and sinuses with an isotonic solution twice daily. Clearing the nose allows children to breathe through their nose, reduces the need to blow their nose or sniff throughout the day, improves sleep quality, improves sense of smell and taste, enables children to eat with a closed mouth and can improve hearing. It is also thought that clearing the mucus might reduce the bacterial load in the nose, preventing passage to the lower airways (Alanin et al 2015).
Children who are too young to perform nasal douching may be offered sea water nasal sprays or nasal drops with nasal aspiration. Anecdotally, parents find baby nasal aspirators help clear the nose and aid sleep in infants.
In addition, children may require nasal steroids, antihistamines, oral or nebulised antibiotics to manage symptoms, but there is no evidence of general benefit and these should be trialled on a case-by-case basis. One study demonstrated that nasal surgery can aid clearance of bacteria from the sinuses and improve quality of life (Alanin et al 2017). Despite chest symptoms being considered the most important aspect of PCD management by healthcare professionals, it is often the nasal symptoms that patients find most bothersome. Children often report bullying in school due to a constant runny nose, eating with their mouth open and sniffing.
Finally, if obstructive sleep apnoea is evidenced by history, sleep study and examination, a tonsillectomy with or without adenoidectomy may be indicated (Lucas et al 2014).
Cardiac care is not commissioned as part of the PCD service. A proportion of PCD patients have other comorbidities owing to heterotaxy or other associated diagnoses. These include malrotation of the gut, asplenia, kidney problems, biliary atresia, Joubert syndrome, retinitis pigmentosa, learning disabilities and immunology problems. Good communication between specialties is essential to ensure that patients' respiratory needs are taken into consideration especially around surgery.
As with all chronic conditions, a good transition is essential to ensure young people are fully supported and empowered to take care of their own health needs going into adulthood (Nagra et al 2015). Currently there is no funded adult service for PCD in the UK. PCD is a rare disease unknown to many medical or nursing professionals, which means that patients may be mismanaged in adult services. Ensuring young people have the knowledge to become ‘expert patients’ with a good understanding of PCD management and skills to negotiate with physicians may serve to reduce morbidity.
All four PCD centres use the Ready Steady Go programme as a framework for transition to adult care. The process starts at 11 years old when children are moving to senior school, thereby taking more responsibility for all aspects of their lives. The process is gradual, covers all aspects of daily life and builds on children's knowledge and skills until they are ready to transfer to adult services at 16-18 years old. Alongside this, children with a diagnosis of PCD are given a ‘PCD folder’ from diagnosis. This has sections including diagnostic information, PCD Family Support Group information and letters/reports for audiology, cardiac, annual review, general review, discharge summaries, school and disability living allowance (DLA). The folder ensures that information about the child is kept in one place should the child need to be seen in a non-PCD centre. It also means that when young people need to take responsibility for their health, they have a comprehensive record of their journey.
Go to uhs.nhs.uk/Ourservices/Childhealth/TransitiontoadultcareReadySteadyGo/Transitiontoadultcare.aspx Discuss with your colleagues the implications of being a young person diagnosed with a rare genetic disease. PCD has implications for fertility (not discussed in this article) and, as a genetic condition, it may be passed down to the next generation especially in populations where interfamily marriage is common. Discuss the possible effects a poor transition may have on a young person with a chronic condition. Consider adherence to treatment regimens and outpatient clinic attendance.
Ready Steady Go offers a framework to discuss these difficult and complex issues while remaining in a safe environment.
The primary role of the PCD clinical nurse specialist is to facilitate management of the child in the community, reduce the number of hospital admissions and ensure children can reach their full potential by communicating with community agencies and schools to ensure all the child's needs are met.
Each centre offers a telephone support service that may triage patients, recommend tests, prescribe antibiotics and offer advice to patients, families and other professionals. Patients requiring review can be directed to the GP, local hospital or specialist service as appropriate. Most children with PCD have open access arrangements with their local hospital allowing them to bypass the GP or emergency department if required. Excellent communication between the PCD team, the family and local services is essential to ensure best outcomes for the child.
Local and regional clinics are coordinated by the team with the clinical nurse specialist taking the lead role. Families may also be supported with claims for DLA and school health plans.
When children do require admission, the clinical nurse specialist works with ward staff and local hospitals to ensure all the child's care needs are met.
Research is an important aspect of the PCD service contributing to an emerging evidence base. Nurse specialists ensure the seamless integration of research into clinical practice. Evidence in PCD is sparse and, as a rare disease, recruiting sufficient patients to power clinical trials requires national and/or international collaboration. The first international randomised controlled trial is nearing completion, having investigated the efficacy of prophylactic azithromycin to reduce respiratory exacerbations (Kobbernagel et al 2016). As clinical trials are proposed, there is a risk that patients will be overburdened with research; the clinical nurse specialist can empower patients to take part in available research but also to refuse if they wish. It is vital that research undertaken is relevant and important to patients. Close collaboration with the PCD Family Support Group helps guide future research projects and balance the needs of an emerging evidence base with the individual burden on the patient.
PCD is rare and underdiagnosed; symptoms need to be recognised by healthcare professionals to improve referrals to the diagnostic service. Early diagnosis and management are likely to reduce morbidity and mortality, delay irreversible lung damage, increase educational achievement and improve patients' quality of life. The disease trajectory is not fully understood due to the lack of research. Increased diagnosis and an emerging evidence base for treatment should improve outcomes for PCD patients. Nurses from many backgrounds, for example, neonatal, respiratory, ear, nose and throat and cardiology, are ideally placed to identify patients for diagnostic referral by considering the history and perhaps using PICADAR (Behan et al 2016a) as a guide for referral.

07 March 2024 / Vol 36 issue 2
How play specialists can reduce use of anaesthesia during radiotherapy
Radiotherapy practice is complex and daunting for children....
Developments in diagnostics and treatment for myeloma
Over the past decade, many novel treatments for myeloma have...
Management of patients with low-risk febrile neutropenia
The National Institute for Health and Care Excellence in the...
View from the front line
In December 2008, readers of RCN Publishing’s eight...
Redesigning the response to patients with trauma
In 2008, a multidisciplinary team of nurses, doctors,...